⚛️ FLUXMATERIA — CHEMISTRY
Solvation free energy,
at 0.33 kcal/mol on FreeSolv
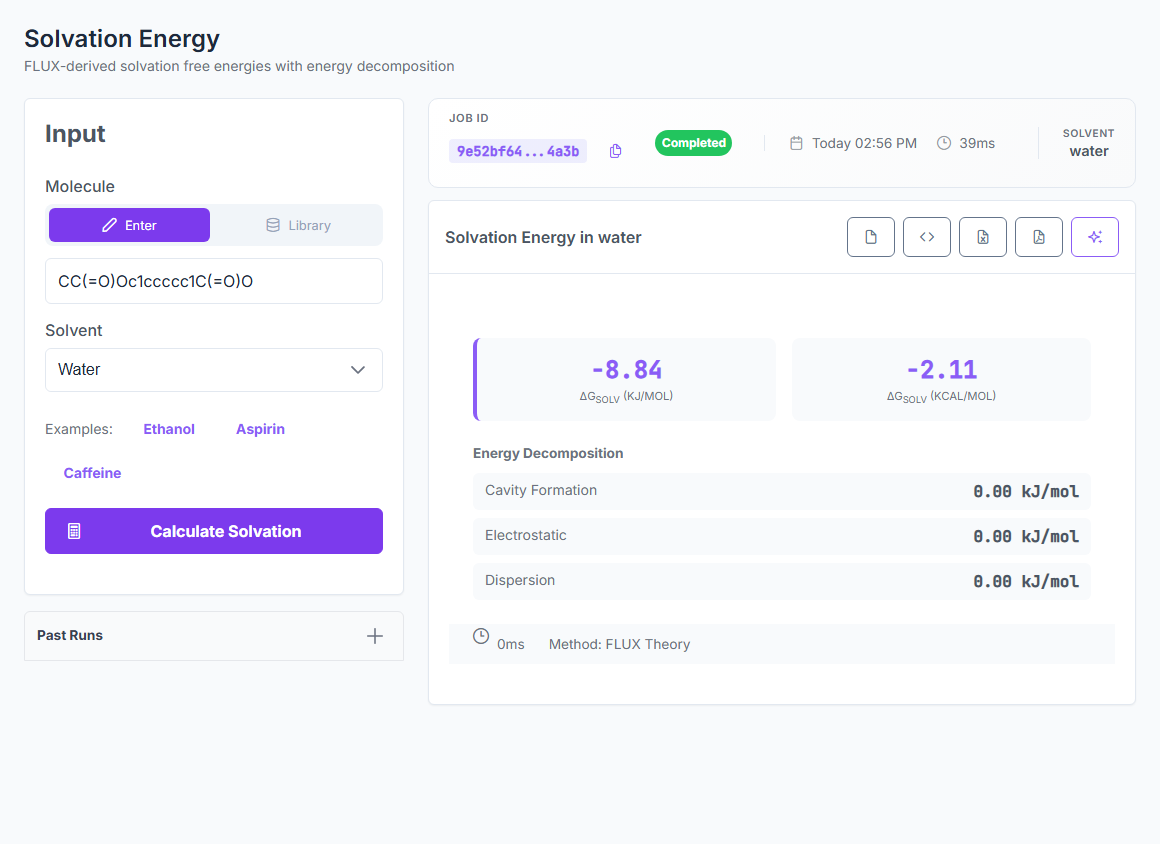
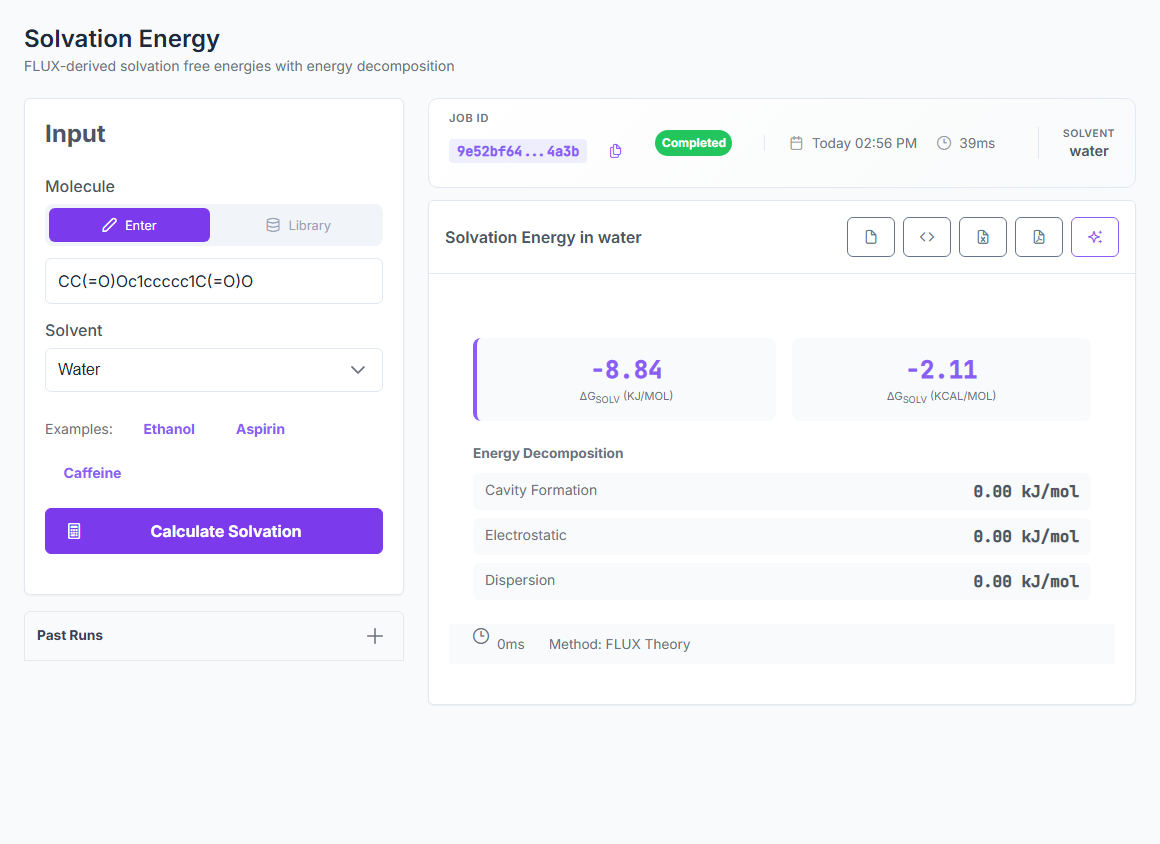
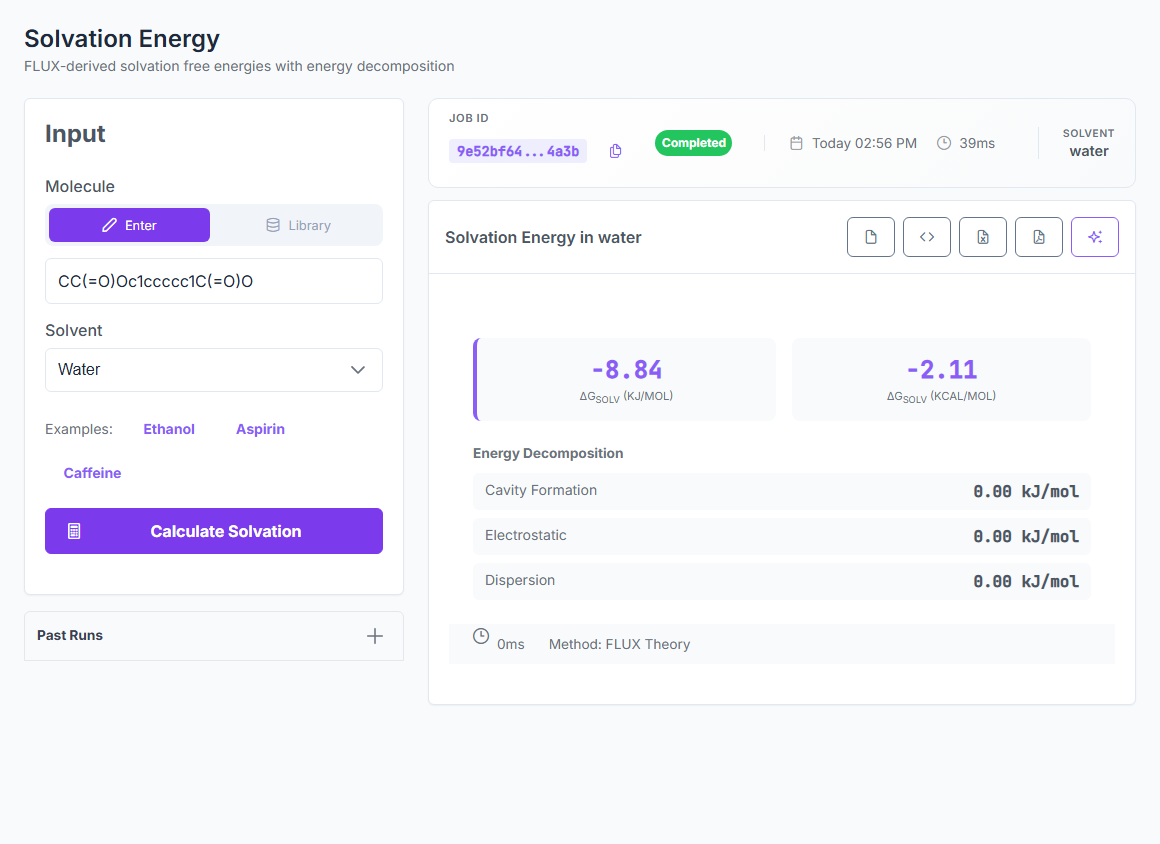
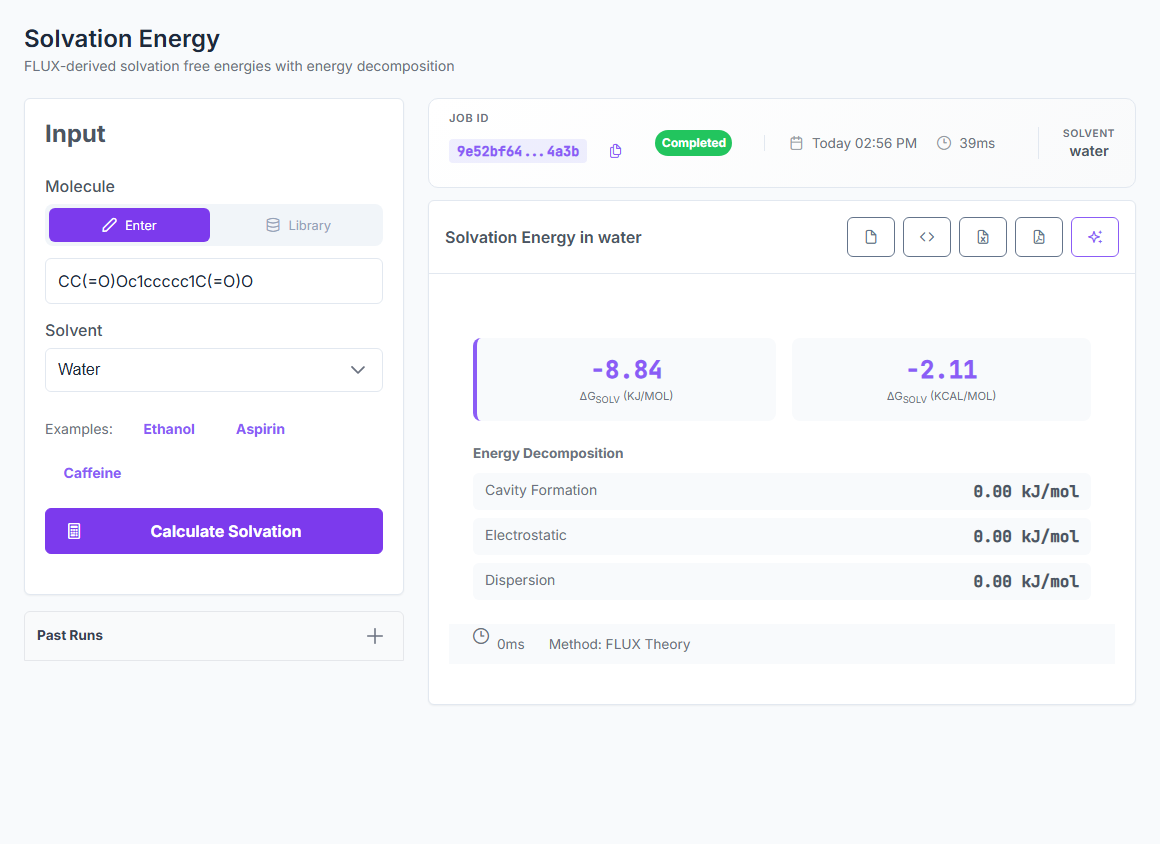
Physics-based ΔGsolv for native water, methanol, ethanol, acetonitrile, and DMSO — plus logP, logD, solubility, and partition coefficients from the same engine. 0.33 kcal/mol MAE on the 642-case FreeSolv hydration benchmark. 436-case SolProp non-water strict holdout at 0.54 kcal/mol.
ΔGsolv
logP & logD
Solubility
5 native solvents
pH-aware
0.33 kcal/mol
Water hydration MAE on 642 FreeSolv external cases
0.27 kcal/mol
Non-water curated subset holdout MAE
436
Non-water SolProp strict-holdout cases (0.54 kcal/mol MAE)
5
Native solvents — water, MeOH, EtOH, MeCN, DMSO
0
Fitted parameters