⚛️ FLUXMATERIA — CHEMISTRY
SN1 / SN2 / E1 / E2 / E1cb,
at ~1 ms per prediction
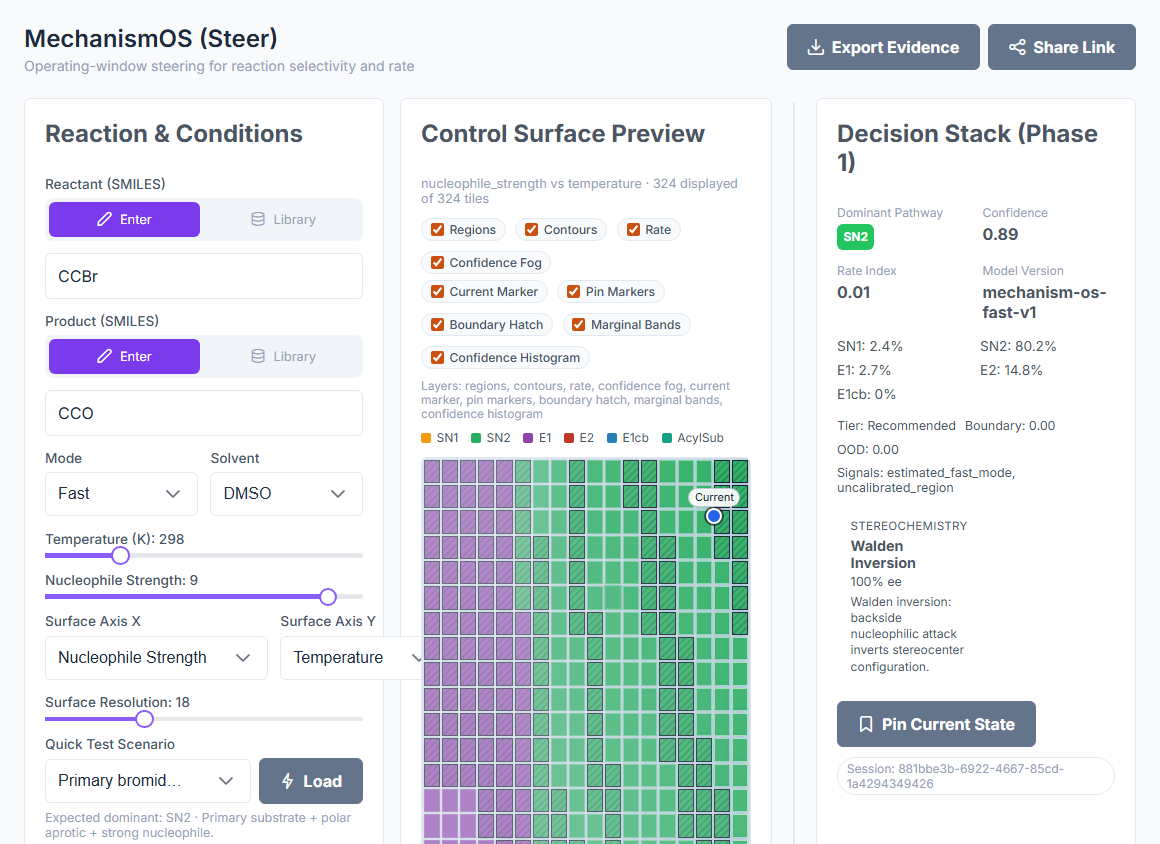
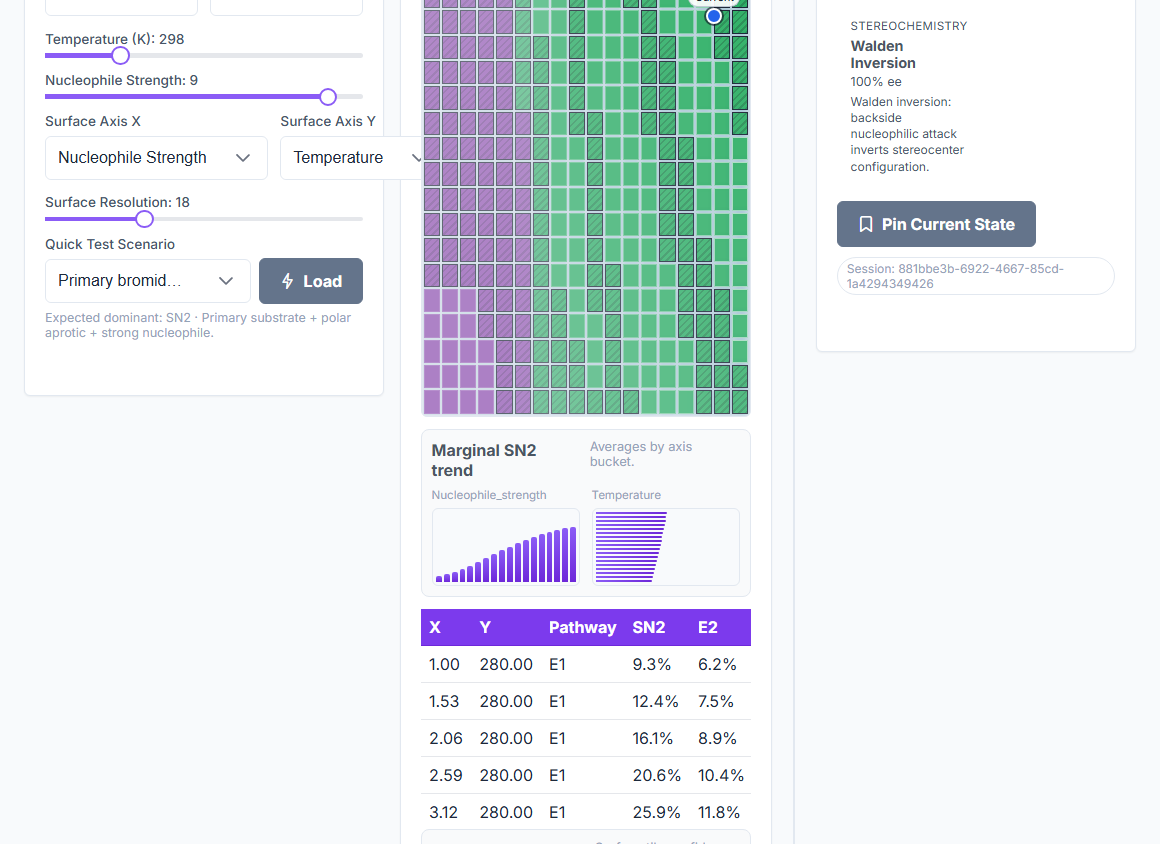
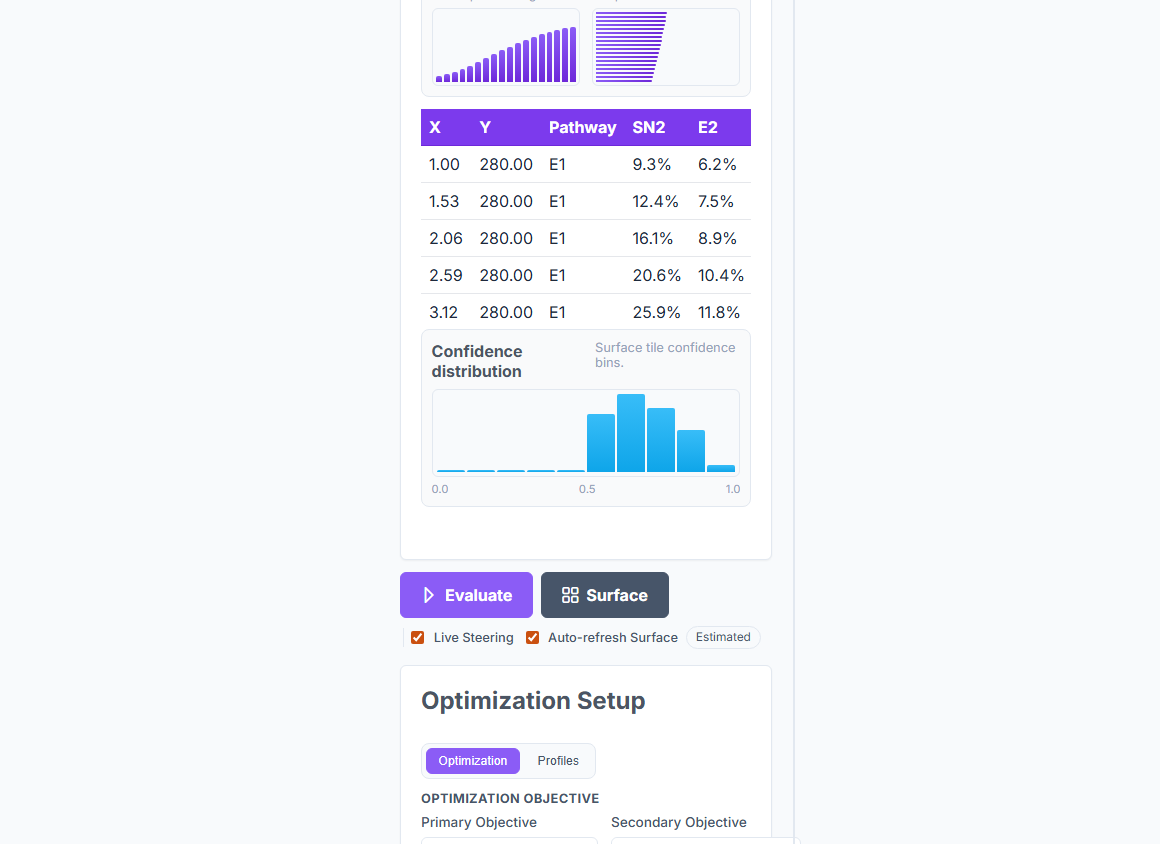
Mechanism Discovery classifies substitution and elimination pathways with 100% accuracy on the 336-case published benchmark, returns the activation barrier at 7.4 kJ/mol MAE, and models temperature crossovers, rearrangements, and neighbouring-group participation — in about a millisecond per prediction.
5 mechanisms
16 solvents
7 leaving groups
Rearrangements & NGP
No ML
100%
Mechanism accuracy on 336 published cases
7.4 kJ/mol
Activation barrier MAE vs experiment
~1 ms
Per-prediction runtime (vs hours for DFT)
106×
Speed-up vs conventional quantum chemistry
0
Fitted parameters