⚛️ FLUXMATERIA — CHEMISTRY
Every molecular property
from a single SMILES
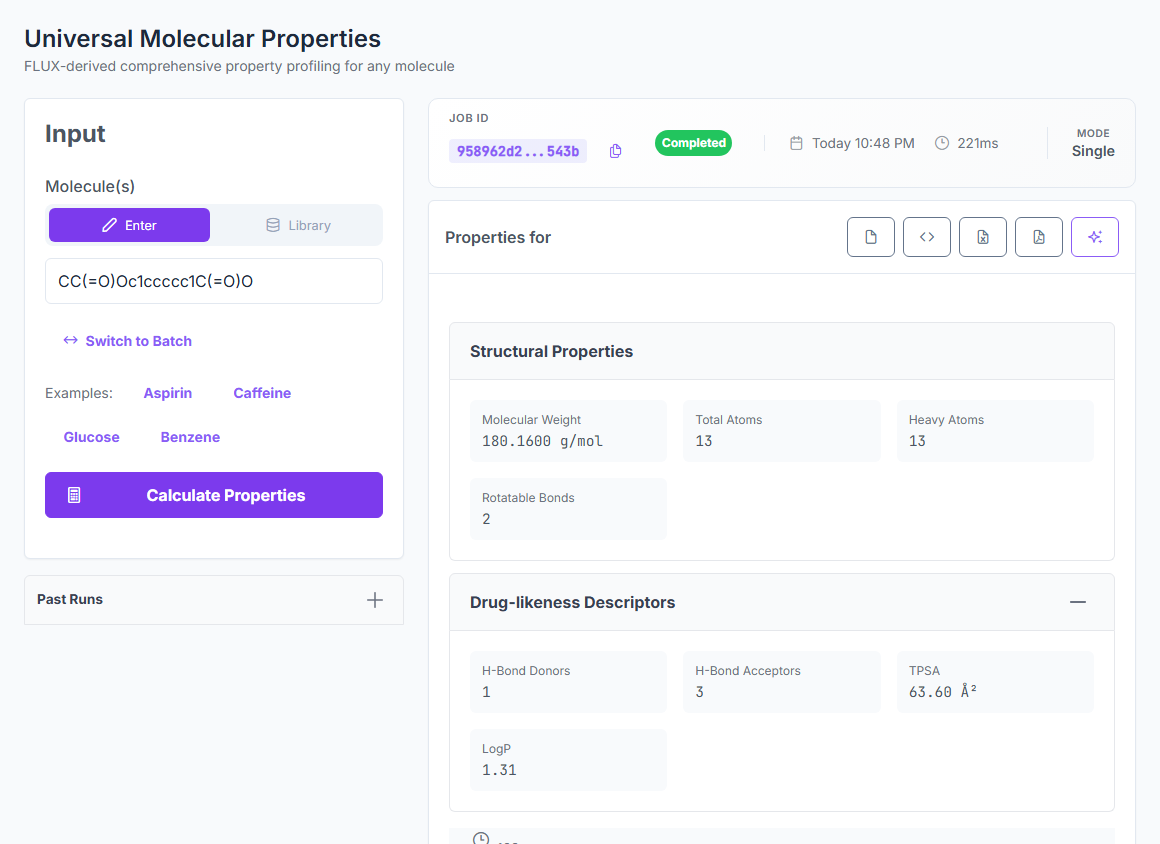
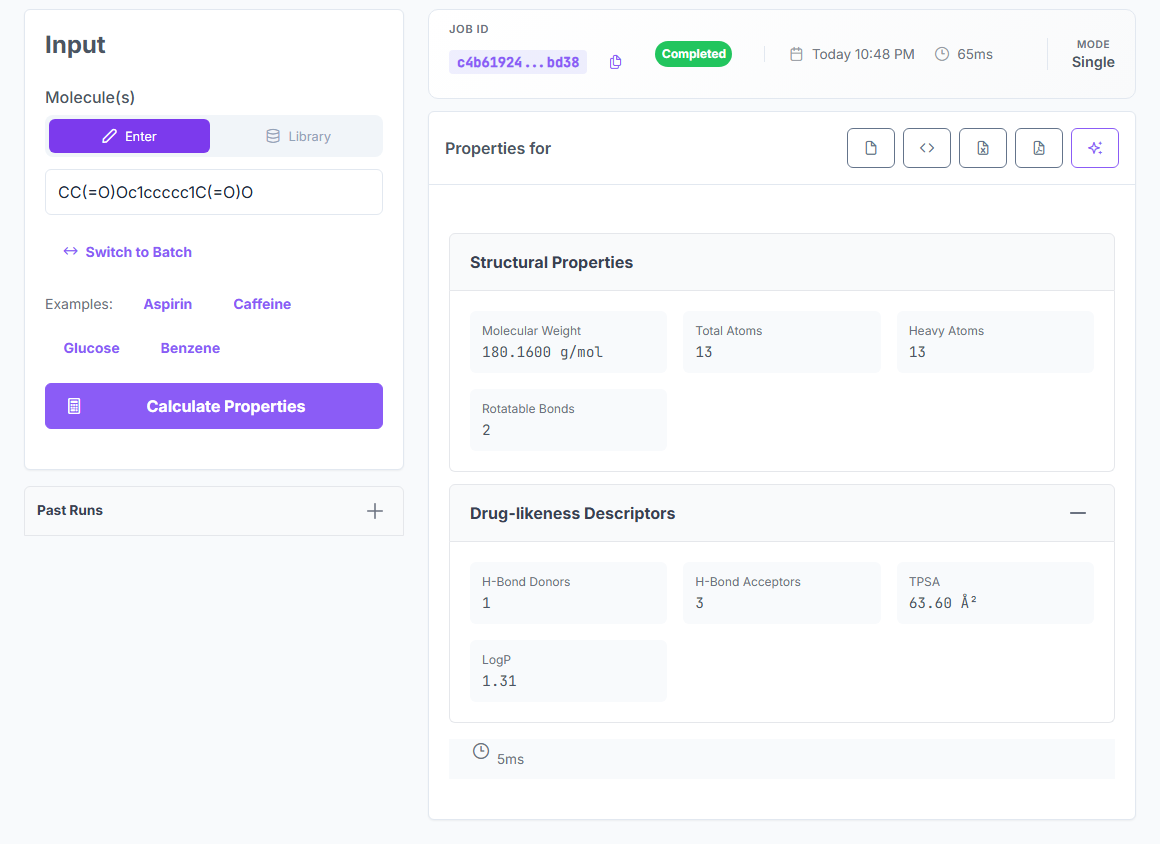
Advanced Methods is the universal calculator. One request returns structural descriptors, drug-likeness, electronic properties, and reactivity indices — all from first-principles physics, all with zero fitted parameters.
Structural
Drug-likeness
Electronic
Reactivity
Batch of 100
5,956
First-principles formulas across 75 JSON layers
0.079%
Bond-length MAPE across 453 validated bonds
0.289%
Bond-energy MAPE across 908 validated BDEs
100
Molecules per batch request
0
Trained parameters